Top menu

The top menu is present at any time and contains icons that provide means to:
- Return to the home-page
- Display a map geolocalizing populations present in the database
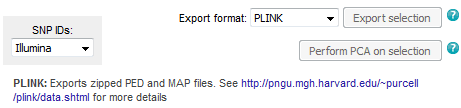
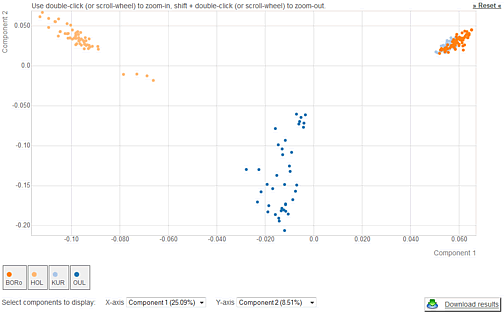
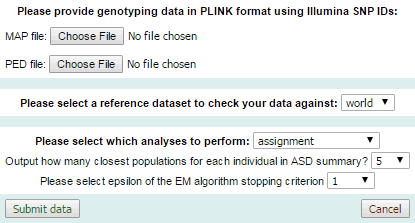
- Upload genotyping data for new individuals, perform PCA with WIDDE individuals and population assignment
- Log in and out
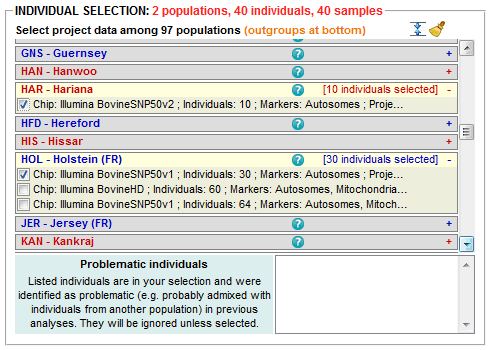
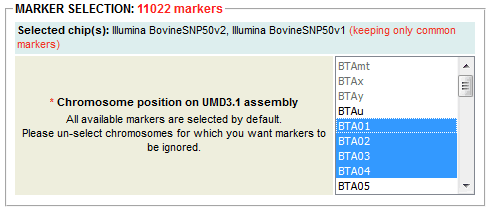
 Batch-select individuals genotyped on one or several SNP chip(s) (Illumina BovineSNP50v1, IlluminaSNP50v2 and Illumina Bovine HD) and individuals that belong to one or several population group(s) (European taurine, African taurine, Zebu, Hybrid and Outgroup)
Batch-select individuals genotyped on one or several SNP chip(s) (Illumina BovineSNP50v1, IlluminaSNP50v2 and Illumina Bovine HD) and individuals that belong to one or several population group(s) (European taurine, African taurine, Zebu, Hybrid and Outgroup) Hide unselected populations
Hide unselected populations Show unselected populations
Show unselected populations